【经验总结】方法开发梯度洗脱问题汇总
梯度洗脱的原理
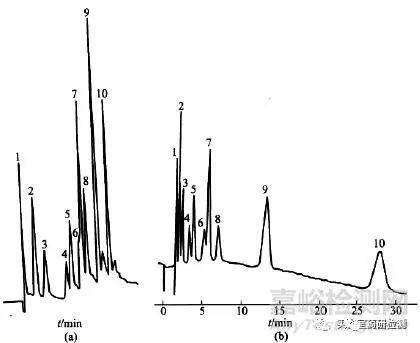
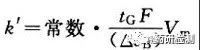
梯度洗脱出现的问题
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
梯度滞后体积
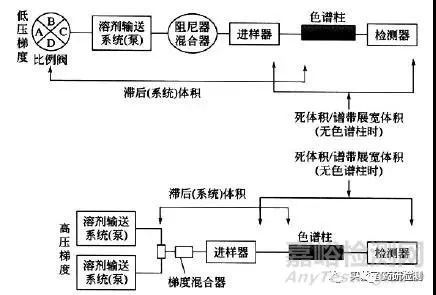
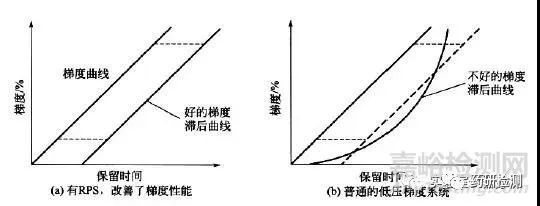
展源
何发
相关文章
-

梯度洗脱 VS 等度洗脱?
2023-02-15
-

梯度洗脱 VS 等度洗脱 ?
2024-06-28
-

方法开发梯度洗脱问题小结
2021-10-20
-
准确度?正确度?精密度?还在为此凌乱吗?
2020-05-27
-

检测误差、检测准确度、检测不确定度,有什么不同?
2023-04-11
-

检测误差、检测准确度、检测不确定度,有什么不同??
2023-11-29
-

4万亿摄氏度高温度下所有物质都将分解为"汤"度
2020-05-27
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-

QC, IQC, IPQC, QA,到底是什么鬼?
2020-05-27

加载更多